Photo by Sangharsh Lohakare on Unsplash
NVIDIA Parabricks is a software suite that accelerates genomic sequence analysis by reimplementing industry standard tools to use the parallel processing power of NVIDIA GPUs. As a result, Parabricks is up to 108x faster than CPU-native variants of the same tools.

These substantial performance improvements speed up time-sensitive sequencing tasks such as identifying what pathogen is causing an infection in order to administer the right treatment. Additionally, GPU acceleration with Parabricks reduces the cost of running bioinformatics-based workloads, boosting accessibility of these important applications. However, increased speed and reduced cost are only valuable if they do not negatively impact accuracy. Parabricks provides better accuracy thanks to high accuracy deep learning for all major sequencers.
Parabricks affords the opportunity for better performance, faster development, and lower costs with equivalent output to gold-standard tools across a range of scientific workflows. Coordinating Parabricks tasks with a modern workflow orchestrator offers additional benefits, such as:
- Defining infrastructure requirements: Parabricks is optimized for use with NVIDIA GPUs. Being able to articulate this in a loosely-coupled and platform-agnostic way accelerates the development lifecycle.
- Capturing heterogeneous dependencies: being able to easily add non-Parabricks dependencies to a container image for auxiliary tasks in a workflow improves reproducibility.
- Dataflow awareness: capturing the inputs and outputs at the boundaries between different Parabricks tasks and annotating them with metadata enhances the reliability and predictability of every execution.
Flyte is an open-source, AI-focused workflow orchestration engine that makes it straightforward to build bioinformatics pipelines. Advanced teams at companies such as Cradle Bio, Gingko Bioworks, and Astra-Zeneca have adopted Flyte to orchestrate and compute scientific pipelines. Bioinformaticians and other practitioners value Flyte’s reliability, reproducibility, and ergonomics, which are achieved through the following features:
- An intuitive Python-based SDK (`flytekit`) allows workflows to be developed in pure Python, expanding accessibility to scientists and other domain experts
- Task-level dependency and infrastructure specification allow for truly heterogeneous workloads to be orchestrated in a single pipeline. This is extremely useful in the world of bioinformatics, where every step in a pipeline uses different tools and data
- Declarative dependency management alongside code via ImageSpec allows teams to quickly iterate on workflows without writing Dockerfiles or manually building containers
- Strongly-typed inputs and outputs and support for complex dataclasses allow for compile-time error checking and highly accurate caching, which can dramatically reduce execution costs and run times
- Accelerator specification and Agents allow users to target specific GPUs and accelerated cloud solutions such as DGX Cloud with minimal code changes
To demonstrate the advantages of orchestrating Parabricks-based workloads with Flyte, we’ve ported the Whole-Genome Small Variant Calling workflow from the Parabricks documentation into a Flyte workflow. Before diving into the example, it’s worth noting some of the complexities involved in the original example:
- Multiple machine types: the example requires the minimum hardware requirement of two NVIDIA V100 GPUs, 24 CPU cores, and 512 GB of disk. “Exotic” dependencies: both `samtools` and `BWA` are C-compiled libraries that assume access to a local file system. The example instructs users to install these locally or write Dockerfiles and manage containers
- Files on disk: the inputs and outputs of the various steps in this workflow are serialized to disk, which can introduce tedious-to-debug errors which are often not easily reproducible
Flyte makes it possible to programmatically declare each step of this workflow in pure Python, removing the need for `docker run` commands and thus boosting reusability, readability and debuggability. Infrastructure (i.e. GPUs, CPUs, and memory) as well as code dependencies are provisioned declaratively at the task level, which allows different steps in the pipeline to leverage different compute resources and environment configurations. The inputs and outputs of each step in the workflow are also serialized using Python dataclasses, and intermediate values are stored automatically in an object store, improving reproducibility.
In summary, Flyte makes it significantly easier for practitioners who know Python to develop powerful bioinformatics pipelines.

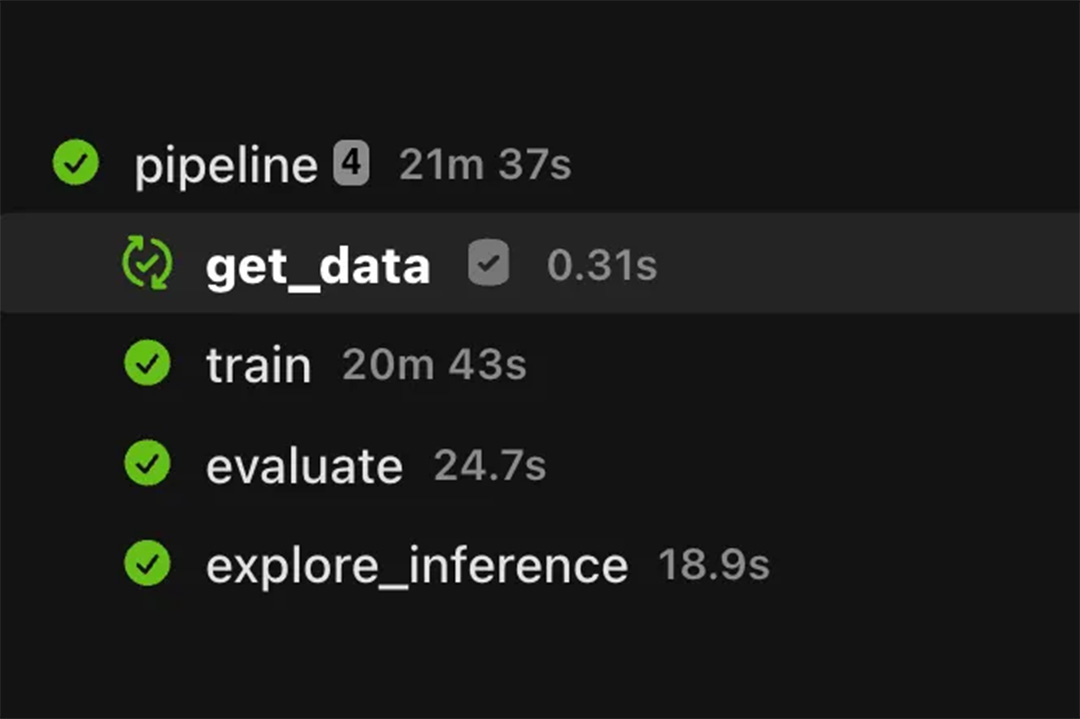
This workflow, visualized above, performs the following steps in order:
- The first few tasks fetch the necessary inputs and generate an index for the reference. Fetching the inputs in this way replaces manual `wget` calls and allows users to cache the workflow inputs, dramatically speeding up subsequent executions.
- At the core of the workflow are the Parabricks tasks (prefixed with `pb`), which consist of an alignment step followed by a calling step across two different callers
- Finally, an intersect task creates the intersection of both caller’s Variant Call Format (VCF) output files, to generate a more sensitive set of variants
Each task in the workflow is manifested as a Kubernetes pod with its own infrastructure, container image, and typed inputs. When the workflow executes, all intermediate inputs and outputs are persisted to an object store and remain accessible in Flyte’s UI. Furthermore, all tasks, workflows, and executions are immutable and automatically versioned, so each execution is reproducible.
Here is the definition of the above Flyte workflow:
Here, the function `wgs_small_var_calling_wf` is annotated with the `@workflow` decorator, telling Flyte that this is a workflow definition. The structure of the DAG is determined by the dependencies between the function calls in the body of the workflow. Flyte will automatically parallelize the three `fetch` commands, since none depends on the output of the other.
While the `fetch` tasks are fairly straightforward requests, they do produce custom dataclasses that serve to organize and enrich the underlying files. For instance, the `Reference` dataclass is defined as follows:
Since different aligners index in different ways and require different index arguments, the dataclass relies on a FlyteDirectory to capture everything, then produces the appropriate string when needed. Bioinformatics tools often assume a local file system and consume inputs based on file extensions or fairly inflexible directory structures. Using a dataclass instead allows users to better reason about the inputs and outputs getting passed between different tools.
Here is the `bwa_index` task that adds an index to the `Reference` dataclass:
The example above highlights the simplicity and strength of Flyte’s programming model:
- Programmatically specify a custom container image that contains the `samtools` and `bwa` dependencies
- Programmatically specify custom resource requirements, for example the bwa_index task may need up to 10GiB in memory
- Enable caching and set a cache version based on the hash of the reference URI, which means users don’t have to recompute this fairly static asset during subsequent development cycles
The Parabricks tasks leverage dataclasses in a similar way. First, we pull in the necessary attributes from the dataclass, then run a shell command, then update or create a new dataclass, and then return it. For example, the below Flyte task uses the Parabricks-accelerated version of DeepVariant, a popular deep learning-based variant caller from Google:
Code is omitted for the `Alignment` and `VCF` dataclasses since the idea is the same as for the `Reference` type. However, a different image is used — `pb_image`. This is the Parabricks base image with `flytekit` layered on top of it for use in the workflow. Bioinformatics tools come in a variety of languages and a wide range of dependencies, often leading to environment conflicts and bloat. Flyte lets you effortlessly compose workflows with heterogeneous images, giving teams flexibility in how they manage dependencies.
Finally, the workflow calls `intersect_vcfs`, a task that wraps `bcftools isec` to create a more sensitive set of variants from the intersection of two or more VCFs. While there isn’t any explicit code for this in the Parabricks guide, this is a fairly standard tool and is easily wrapped in a final Flyte task, the output of which becomes the workflow output.
Orchestrating Parabricks tasks with Flyte presents a powerful and accessible way of running bioinformatics workflows at scale, providing an exponential speedup in going from sample to solution. While this is a fairly straightforward example, the advantages of wrapping these steps as discrete tasks means that users can easily extend this workflow. In order to process 100 samples, users can compose the Parabricks tasks into a subworkflow and map over all the inputs in parallel. Swapping aligners is also straightforward because of Flyte’s strong typing and support for dataclasses. For instance, the `pb_fq2bam` task could easily be swapped for a task that uses `bwa mem` without any other code changes. Flyte provides the flexibility to compose workflows as fast as users can devise new approaches, and Parabricks is a powerful example of integrating the latest hardware acceleration technology to boost performance.
Full code for this and other examples is available in the UnionBio repository, a growing collection of bioinformatics tasks and workflows built on Flyte.
To get started with Flyte, visit flyte.org and join our Slack. For updates on product development, community events, and announcements, follow us on Twitter to join the conversation and share your thoughtsIf you run into trouble, please let us know by creating a GitHub issue. If you find Flyte useful, don't forget to ⭐ us on GitHub.
About the Authors

Pryce is a Solutions Architect at Union.ai focused on supporting biotech customers. After earning a degree in Molecular Biology from the University of Washington, he acquired his software skills through industry roles in bioinformatics. Pryce is passionate about empowering life-sciences researchers from all backgrounds to do their best work at the highest velocity.

John is a Product Manager at Union.ai focused on building Flyte and the Union AI platform. John earned an MSE in Data Science from Penn Engineering and an MBA from Wharton. He worked in quantitative trading as a portfolio manager before moving to startups where he has held roles in business operations, sales engineering, and product management.